















„Durch den individuellen und richtigen Einsatz der zur Verfügung stehenden Hilfsmittel kann jeder Patient ein großes Stück Lebensqualität und Normalität zurückgewinnen. In diesen Dienst stelle ich meine Fachkompetenz.“

„Wir agieren visionär und menschlich. Wählen Sie uns als Partner und sichern sich zufriedene Kunden.“

“Täglich kann ich zur Genesung und zum Wohlbefinden von Menschen beitragen. Ich liebe, was ich tue, und ich tue es mit Leidenschaft. Aus eigener Erfahrung weiß ich, wie schnell eine schlimme Diagnose das Leben verändern kann. Anderen bei der Auswahl des richtigen Hilfsmittels zu helfen, erfüllt mich mit Freude.”


































Mai
15

Ernährung im Alter
Online
15.05.24
Mai
23
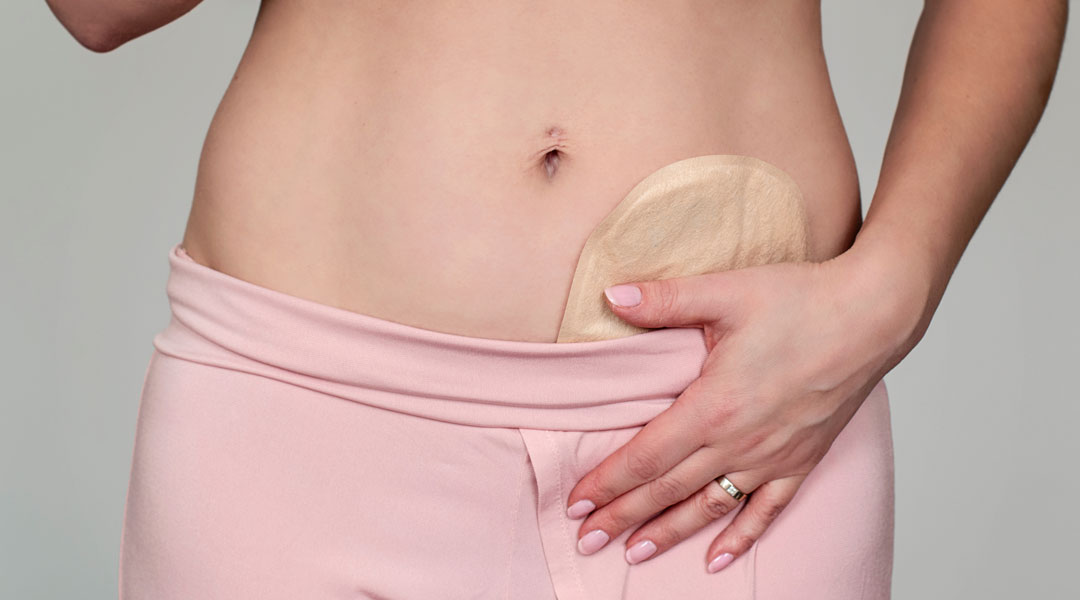
GHD Masterclass: Stoma - Konvexe Stomaversorgung direkt post-operativ!
Online
23.05.24
Mai
31

Palliativ quer durch Hamburg
Goldbach Palliativpflege Team - Jessenstraße 4
22767 Hamburg
22767 Hamburg
31.05.24
Juni
05
Thüringer Onkologie-Nachmittag
GHD GesundHeits GmbH Filiale Erfurt-Stotternheim - Erfurter Landstraße 50
99095 Erfurt-Stotternheim
99095 Erfurt-Stotternheim
05.06.24
Juni
06
Stoma und Wundversorgung
Online
06.06.24
Juni
06
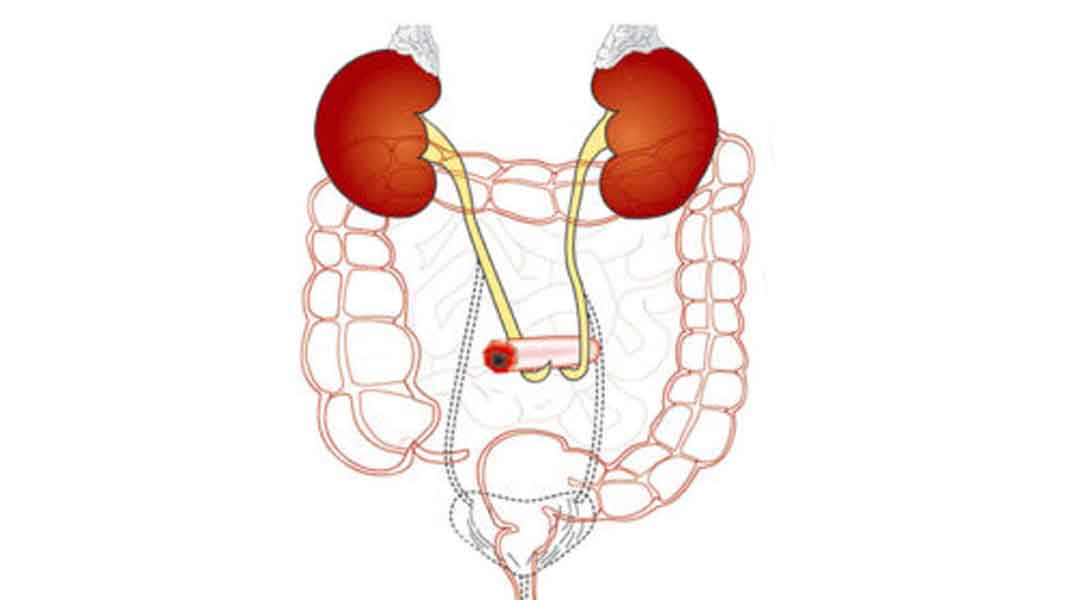
GHD Masterclass - Urostomie-Versorgung
Online
06.06.24
Juni
12

Dresdner Onkologie-Nachmittag
GHD Homecare - Glashütter Straße 53
01309 Dresden
01309 Dresden
12.06.24
Juni
19

Expertenstandard „Schmerzmanagement in der Pflege“
Online
19.06.24
Juni
22

2. Palliativtag in Mecklenburg-Vorpommern
Wohlfahrtseinrichtungen der Hansestadt Stralsund GmbH - Grünhufer Bogen 1A
18437 Stralsund
18437 Stralsund
22.06.24
Juni
26
Port-Workshop in Erfurt
GHD GesundHeits GmbH Filiale Erfurt-Stotternheim - Erfurter Landstraße 50
99095 Erfurt-Stotternheim
99095 Erfurt-Stotternheim
26.06.24
Juli
10
Diabetes und Ernährung
Online
10.07.24
Juli
11

GHD Masterclass: Stoma - Parastomales Pyoderma gangraenosum
Online
11.07.24
August
22
GHD Masterclass: Stoma - Parastomale Hernien- Prävention
Online
22.08.24
September
11

Förderung der Kontinenz in der Pflege
Online
11.09.24
September
18

Ernährung bei Kurzdarmsyndrom
Online
18.09.24
Oktober
08

GHD Masterclass: Stoma - LARS-= Lower anterior resection syndrom
Online
08.10.24
Oktober
16

Dekubitusprophylaxe bei chronischen Wunden
Online
16.10.24
Oktober
22

GHD Masterclass: Der neue "Expertenstandard zur Erhaltung und Förderung der Hautintegrität“
Online
22.10.24
November
06

Mangelernährung in der Dialyse
Online
06.11.24
November
20
Diabetisches Fußsyndrom
Online
20.11.24
November
27
Pflege von Menschen mit chronischen Wunden
Online
27.11.24
Dezember
12

GHD Masterclass: Stoma - Stoma und Ernährung
Online
12.12.24


